

Abstract
Single copy deletions of IKZF1, which occur in 10-15% of all B cell precursor acute lymphoblastic leukemia (BCP-ALL) cases, are associated with a poor outcome. We previously showed that loss of IKZF1 dictates resistance to glucocorticoids (GC) in BCP-ALL cell lines, a knockout mouse model and ex-vivo analysis of primary leukemic cells. When we analyzed the initial response to prednisolone therapy, we found that pediatric patients who suffer from an IKZF1 deleted leukemia are strongly enriched in the poor responder group (14 vs 7%, p<0.05). Although we were able to show that glucocorticoid receptor (GR)-mediated transcription regulation is attenuated in IKZF1 deleted cells, in remained unclear how loss of IKZF1 affects GR signaling.
In T-ALL, GC resistance is frequently induced by genomic mutations that result in hyperactivation of AKT. AKT mediated phosphorylation of the GR results in suppression of GR function and resistance to GC treatment. Although the genomic loci that are frequently mutated in T-ALL resulting in hyperactivation of AKT, such as PTEN, are unaffected in BCP-ALL, we tested whether aberrant activation of AKT may also cause resistance to GC in BCP-ALL. Indeed, both CRISPR/Cas9 induced knockout and shRNA mediated knockdown of IKZF1 resulted in activation of AKT in human BCP-ALL cell lines. This hyperactivation was also observed in splenic B cells isolated from Ikzf1+/- mice. Active AKT in turn promotes phosphorylation of the GR on Ser143. This phosphorylation inhibits GR function, at least in part, by preventing shuttling of the ligand bound protein to the nucleus.
In order to understand how a single copy loss of IKZF1 activates AKT, we tested expression of upstream regulators of AKT activity and found a near complete loss of PTEN expression, the phosphatase that inhibits PI3K and AKT signaling. Indeed, shRNA mediated knockdown of PTEN in wildtype cells resulted in activation of AKT and induced resistance to GCs. Furthermore, in IKZF1+/- cells we observed an upregulation of HES1, a transcriptional factor that is known to repress expression of the PTEN gene. Of note, HES1 was previously identified as a target of IKZF1 mediated transcriptional repression in erythroid and T cells.
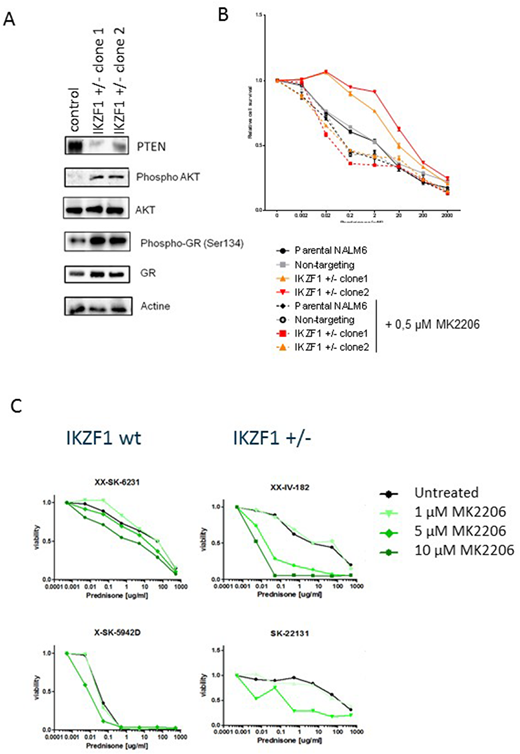
To test whether this AKT mediated mechanism of GC resistance could be reverted through pharmacological inhibition, we tested the effect of AKT inhibitors on prednisolone induced apoptosis of human BCP-ALL cell lines (figure A). In contrast to control cells, where inhibition of AKT had little to no effect on the sensitivity to GC, in IKZF1 deleted cells AKT inhibition reverted the resistance phenotype to a level comparable to control cells. Importantly, also in patient derived xenografts we observed that AKT inhibition was able to sensitize cells to GC treatment (Figure B).
In conclusion, we identified hyperactivation of AKT, as a result of aberrant HES1 expression, as the mechanism causing GC resistance in IKZF1 deleted BCP-ALL. The availability of AKT inhibitors that are under clinical evaluation, may allow the development of combination therapies that restore the response to GCs in IKZF1-deleted BCP-ALL.
Figures:
A) IKZF1, PTEN, AKT, pAKT(Ser473), pGR(Ser134) and GR protein expression levels of NALM6 IKZF1+/- cell lysates were analyzed by western blot. Non-targeting controls were used as control and actin was used as a loading control.
B) NALM6 parental, non-targeting controls or IKZF1+/- cells were treated for 48 hours with increasing concentrations of prednisolone with or without 0.5µM AKT inhibitor MK2206 and analyzed using an MTT based viability assay. All values were normalized to untreated NALM6 cells. Error bars represent ± standard error of the mean (SEM).
(C) hTERT immortalized MSCs were seeded in a 96 wells format and allowed to settle for 24 hours prior to the addition of BCP-ALL patient-derived xenografts (PDX). PDX cells were allowed to settle for 24 hours before prednisone was added in increasing concentrations with or without AKT inhibitor MK2206. After 3 days, sensitivity to treatment was measured by flowcytometry using an amine staining to test for membrane integrity as a measure of cell viability.
No relevant conflicts of interest to declare.
This icon denotes a clinically relevant abstract

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal